
Medycyna mitochondrialna wychodzi z założenia, że wiele chorób jest skutkiem defektów w metabolizmie komórek, a w szczególności uszkodzeń mitochondriów. Ta nowa forma terapii jest stosowana w wielu dziedzinach medycyny, a w ostatnich latach stała się jej głównym obszarem zainteresowania. Pozwala w większym stopniu wpływać na zaburzony metabolizm energetyczny komórek i rozregulowany układ antyoksydacyjny pacjenta. Najważniejszym narzędziem medycyny mitochondrialnej są substancje mitotropowe, za pomocą których podejmuje się próby kompensacji istniejących dysfunkcji. Niniejszy artykuł odnosi się zarówno do substancji mitotropowych, jak i towarzyszących im badań potwierdzających ich skuteczność. Wykazano, że działanie wielu substancji mitotropowych opiera się na dwóch ważnych cechach: po pierwsze, na ich właściwościach przeciwutleniających, gdyż działają bezpośrednio jako przeciwutleniacze oraz aktywują enzymy i szlaki sygnałowe układu przeciwutleniającego, a po drugie, zwiększają transport elektronów i protonów w mitochondrialnym łańcuchu oddechowym.
1. Wstęp
Średnia wielkość mitochondriów wynosi zaledwie 0,75-3 µm, co sprawia, że są one jednymi z najmniejszych organelli w naszych komórkach, a mimo to stanowią około 36% objętości komórek mięśnia sercowego1. Liczba mitochondriów w komórce różni się w zależności od typu komórki i może ulegać zmianom w zależności od zapotrzebowania komórki na energię. Na przykład, w pojedynczej komórce mięśnia sercowego pracuje kilka tysięcy mitochondriów, podczas gdy w dojrzałej komórce jajowej nawet 100 0002. W zależności od ich wagi, wytwarzają o 10 000 – 50 000 razy więcej energii niż słońce – energii w postaci adenozynotrifosforanu (ATP)3. Oprócz produkcji energii, mitochondria pełnią również różne inne funkcje w komórce i są zaangażowane między innymi w regulację reakcji redukcji i utleniania (redoks), proliferację komórek, syntezę hemu i sterydów oraz apoptozę4. Zatem oczywiste jest, że zaburzenia w funkcjonowaniu tych organelli, czy to w wyniku mitochondriopatii, czy też ograniczenia funkcji związanego z wiekiem, mają istotny negatywny wpływ na sprawne funkcjonowanie procesów komórkowych. W poniższym przeglądzie, w celu lepszego zrozumienia, w pierwszej kolejności omówimy funkcjonowanie mitochondriów, a następnie przejdziemy do naturalnych substancji mitotropowych i ich wpływu na układ komórkowy jako potencjalnej korzyści w medycynie mitochondrialnej.
1.1. Funkcja i genetyka mitochondriów
Mitochondria (Rycina 1) to proteobakterie aerobowe, które prawdopodobnie powstały na drodze endosymbiozy (teoria endosymbionta) i występują w prawie wszystkich komórkach eukariotycznych. Uważa się, że endosymbioza jest przyczyną powstania dwóch dwuwarstw lipidowych otaczających mitochondria, określanych mianem błony zewnętrznej i wewnętrznej.
Błona zewnętrzna całkowicie otacza mitochondrium, zamykając jednocześnie przestrzeń międzybłonową. Ma ona wyższą przepuszczalność niż błona wewnętrzna. Zawiera poryny, które są przepuszczalne dla małych cząsteczek i jonów o wielkości do 5 kilodaltonów (kDa)5. Przestrzeń międzybłonowa jest częścią nieplazmatyczną, pełniącą ograniczone funkcje. Błona wewnętrzna przylega do macierzy i tworzy grzebienie, których liczba różni się w zależności od typu komórki. Grzebienie znacznie zwiększają powierzchnię błony, pozwalając na zwiększoną produkcję adenozynotrójfosforanu (ATP) poprzez znajdujący się tutaj łańcuch transportu elektronów (ETC) i syntetazy ATP6. Pomiędzy cytoplazmą a macierzą zachodzi intensywna wymiana metabolitów, dlatego błony mitochondrialne zawierają wiele białek transportowych, takich jak TIM (translokaza błony wewnętrznej) i TOM (translokaza błony zewnętrznej). Jednocześnie błony pozostają nieprzepuszczalne dla wielu jonów i cząsteczek. Matryca zawiera liczne rybosomy, mitochondrialne DNA (mtDNA) (w maksymalnie dziesięciu identycznych kopiach) i ziarnistości. Zachodzą w niej wszystkie główne procesy metaboliczne mitochondriów, w tym replikacja, transkrypcja i translacja genomu5. Jak wspomniano wyżej, mitochondria mają szeroki zakres zadań i oprócz generowania ATP odgrywają centralną rolę w programowanej śmierci komórki (apoptozie) poprzez uwalnianie cytochromu c, spowodowane zmianą przepuszczalności błony mitochondrialnej. W ekstraktach bezkomórkowych wykazano, że inicjacja kaspaz jest możliwa tylko w obecności cytochromu c i dATP (trifosforanu deoksy-adenozyny)7. Śmierć komórkowa jest także wywoływana przerwaniem metabolizmu energetycznego za pośrednictwem łańcucha transportu elektronów8. Za podstawową funkcję mitochondriów uważa się syntezę klastrów żelazowo-siarkowych (klastrów Fe-S). Klastry Fe-S są kofaktorami w różnych reakcjach enzymatycznych, a zatem mają ogromne znaczenie dla przetrwania komórki9.
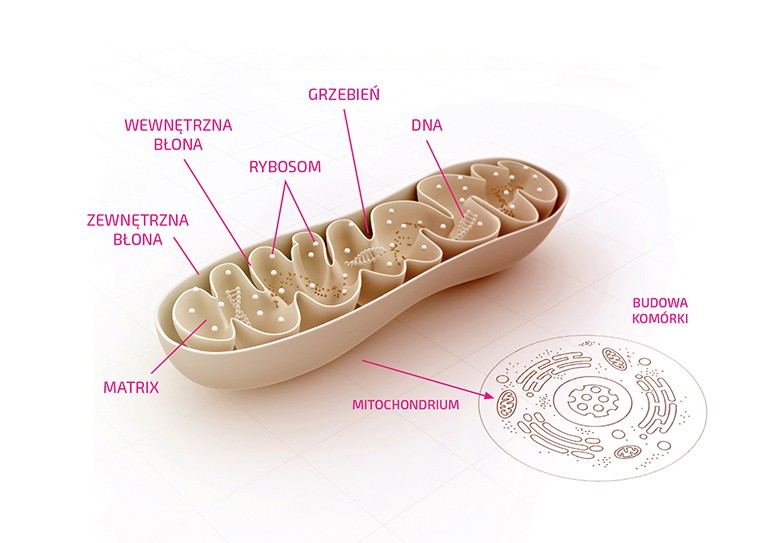
Rycina 1. Schematyczna struktura mitochondrium. Mitochondria są otoczone dwiema błonami definiującymi przestrzeń międzybłonową i macierz. Wewnętrzna błona jest silnie pofałdowana i tworzy grzebienie, co zwiększa jej powierzchnię. Mieszczą się tu kompleksy łańcucha transportu elektronów (ETC). Obie błony mitochondrialne zawierają dużo białek transportowych, np. TIM (translokaza błony wewnętrznej) i TOM (translokaza błony zewnętrznej). Macierz zawiera między innymi mitochondrialne DNA (mtDNA), rybosomy i ziarnistości.
Genom mitochondrialny o wielkości 16,5 kilobaz (kb) jest dwuniciowy, kolisty i nie zawiera histonów ani intronów. W 37 genach koduje 2 specyficzne dla organelli rybosomalne kwasy rybonukleinowe (rRNA) o wielkości i sekwencji podobnej do bakteryjnego rRNA, 22 transportujące RNA (tRNA) i 13 niezbędnych białek mitochondrialnych2,10. W mitochondriach znaleziono kilka odmiennych kodonów: u ludzi mitochondrialny kodon AUA koduje metioninę zamiast izoleucyny; AGA i AGG nie kodują argininy, jak to zwykle ma miejsce, tylko są kodonami „stop”. Te różnice między genomem jądrowym i mitochondrialnym uzasadniają pogląd, że mitochondria w komórkach eukariotycznych były pierwotnie symbiontami pochodzenia prokariotycznego, zanim przekształciły się w obowiązkowe składniki komórek eukariotycznych. „Uniwersalność kodu genetycznego” jest jednym z głównych praw biologii molekularnej. Zgodnie z jego zasadą, informacje zawarte w sekwencji nukleotydów są odczytywane i transkrybowane w ten sam sposób przez wszystkie żywe organizmy. Natomiast mitochondria stanowią godny uwagi wyjątek, ponieważ są na ogół półautonomicznymi organellami i mają genom, który koduje tylko kilka białek mitochondrialnych. Tak więc większość czynników regulacyjnych i białek strukturalnych jest kodowana przez jądrowe DNA i musi być transportowana z cytoplazmy do mitochondriów po ich syntezie. Nawet części błony mitochondrialnej i wiele potrzebnych enzymów jest kodowanych w jądrze, więc muszą być importowane do organelli. W związku z tym przez długi czas zakładano, że mitochondria nie są w stanie funkcjonować bez jądra komórkowego. Jednak obecnie wiadomo, że sprawne mitochondria znajdują się nie tylko w bezjądrowych płytkach krwi, ale mogą również występować w dużych ilościach we krwi jako mitochondria bezkomórkowe11. Podczas podziału komórki mitochondria przechodzą do komórek potomnych i rozmnażają się przez podział, który zachodzi autonomicznie i niezależnie od komórki. Mitochondria są dziedziczone po matce, co zapobiega rekombinacji mtDNA, np. podczas procesu mejozy2.
1.2. Łańcuch oddechowy
Wytwarzanie ATP na drodze fosforylacji oksydacyjnej, która zachodzi w wewnętrznej błonie mitochondrialnej, odbywa się dzięki transportowi elektronów wzdłuż kompleksów I do IV oraz mobilnych składników ubichinonu (CoQ) i cytochromu c ETC (Rycina 2). Te dwa ostatnie służą jako czynniki przenoszące elektrony i protony między wspomnianymi kompleksami. Największy kompleks enzymatyczny łańcucha oddechowego, kompleks I, znany również jako oksydoreduktaza NADH-ubichinon, katalizuje transport dwóch elektronów z dinukleotydu 1,4-dihydronikotynamidoadeninowego (NADH) do CoQ oraz transport czterech protonów (H+) z macierzy do przestrzeni międzybłonowej (cykl Q), co przyczynia się do powstawania gradientu protonowego w obrębie błony wewnętrznej. W rezultacie powstaje zredukowany ubichinol (CoQH2), dwuelektronowy nośnik, który może swobodnie dyfundować w dwuwarstwie lipidowej błony wewnętrznej5.
W kompleksie II (dehydrogenaza bursztynianowa), będącym również częścią cyklu cytrynianowego, zachodzi utlenianie bursztynianu do fumaranu, co prowadzi do przeniesienia elektronów do CoQ, w wyniku czego powstaje CoQH2. Kompleks II, oprócz trzech klastrów żelazowo-siarkowych (klastrów Fe-S), posiada grupę prostetyczną w postaci dinukleotydu flawinoadeninowego (FAD) oraz miejsce wiązania bursztynianu. Elektrony przemieszczają się od bursztynianu do CoQ przez FAD i klastry Fe-S5. Następnie w dimerycznym kompleksie III, kompleksie oksydoreduktazy ubichinon cytochrom c, następuje przeniesienie elektronów z CoQH2 na cytochrom c wraz z translokacją czterech protonów z macierzy do przestrzeni międzybłonowej. Cytochrom c, jako nośnik jednoelektronowy, przenosi elektrony z kompleksu III do kompleksu IV i przekazuje je dwóm dwuwartościowym jonom miedzi (Cu2+) oksydazy cytochromu c (CuA) zlokalizowanej w podjednostce II. Podjednostka ta posiada dwie grupy hemowe (hem a) jako grupę prostetyczną5. Kompleks IV, oksydaza cytochromu c, katalizuje transport elektronów z cytochromu c do tlenu (O2) i redukuje go do dwóch cząsteczek wody (H2O). Podjednostka I kompleksu IV posiada dwie grupy hemowe (hem a i hem a3) oraz dodatkowy jon Cu2+ (CuB). Hem a3 i CuB tworzą kolejne centrum dwujądrowe. Tak więc transport elektronów z cytochromu c do O2 odbywa się za pośrednictwem centrum CuA, hemu a i centrum hem a3-CuB. Energia uwalniana podczas redukcji O2 do H2O jest wykorzystywana do pompowania dodatkowych czterech H+ na cząsteczkę tlenu z macierzy przez kompleks IV i błonę wewnętrzną do przestrzeni międzybłonowej. Aby zapobiec powstawaniu reaktywnych form tlenu (ROS) w tym procesie (patrz infografika), centrum hem-a3 CuB musi zostać najpierw zredukowane za pomocą dwóch elektronów. Tylko w takich warunkach możliwe jest wiązanie O2. W ten sposób tlen może zostać zredukowany bezpośrednio do nadtlenku, zanim zostanie rozbity na poszczególne atomy.
Gradient protonowy powstały podczas transportu elektronów katalizuje syntezę ATP z difosforanu adenozyny (ADP) i fosforanu nieorganicznego (Pi) poprzez strumień protonów z przestrzeni międzybłonowej z powrotem do macierzy mitochondrialnej za pomocą syntazy F0F1-ATP (kompleks V). W tym procesie musi być utrzymywane wysokie napięcie elektryczne w obrębie błony mitochondrialnej przy natężeniu pola około 1 000 000 woltów/cm. Wewnętrzna błona mitochondrialna działa jak izolator, podczas gdy w powietrzu iskra elektryczna przeskoczyłaby już przy 10 000 V/cm, a tym samym doprowadziłaby do zwarcia12. Syntaza ATP w mitochondriach jest syntazą ATP typu F i składa się z dwóch komponentów: F1 i F0. F1 jest obwodowym białkiem błonowym z miejscami wiązania nukleotydów, podczas gdy F0 jest zintegrowany z błoną i ma kanał protonowy5. Podjednostka F0 składa się z trzech głównych podjednostek a, b i c oraz sześciu innych podjednostek. F1 jest heksamerem składającym się z trzech podjednostek α i trzech podjednostek β. F1 i F0 są połączone obwodowym i centralnym ramieniem składającym się z podjednostek γ-, δ- i ε. Podjednostka γ doprowadza do kontaktu podjednostek c F0 z podjednostkami δ- i ε. Zgodnie z modelem chemiosmotycznym, siła napędowa protonów napędza syntazę ATP. Powracające protony powodują ruch obrotowy pierścienia podjednostki c F0 i podjednostek γ, δ i ε centralnego ramienia. Powstały ruch obrotowy jest przenoszony na cząsteczkę F1. Powoduje to tworzenie ATP w części F1 poprzez zmianę konformacyjną. W ten sposób ramię obwodowe zapobiega obracaniu się całej podjednostki F1. Gdy podjednostka γ obraca się całkowicie, powstają trzy ATP5. Transfer elektronów i syntaza ATP są ze sobą sprzężone i nie działają niezależnie od siebie13. H+ może być również pompowany z powrotem do macierzy przez inne białka błony mitochondrialnej zwane białkami rozprzęgającymi (UCP). Prowadzi to do odłączenia łańcucha transportu elektronów od syntazy ATP14.
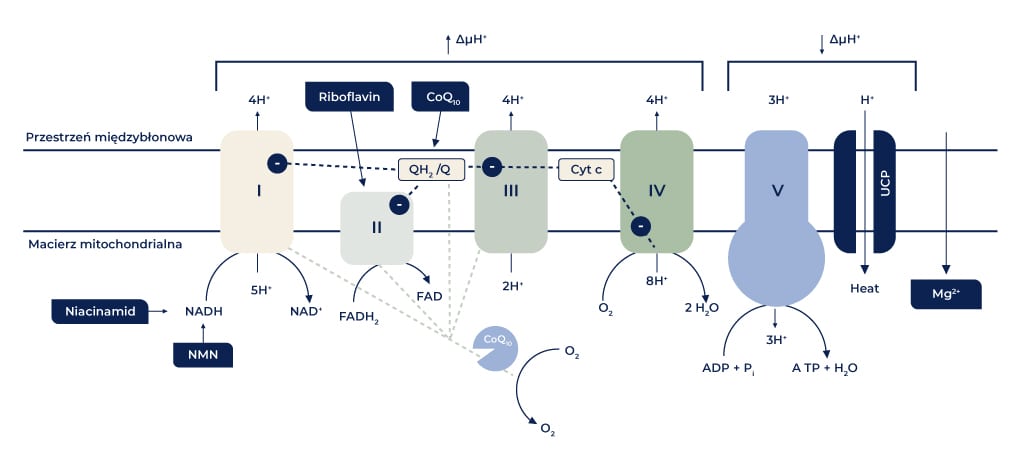
Rycina 2. Uproszczona reprezentacja mitochondrialnego łańcucha oddechowego i celów wybranych substancji mitotropowych. Pokazano wewnętrzną błonę mitochondrialną i przedziały łańcucha oddechowego. Kompleksy I-IV generują gradient protonów (↑∆µH+), dzięki czemu ATP może być syntetyzowane przez kompleks V (syntaza ATP) (↓∆µH+). Elektrony przemieszczają się z kompleksu I przez kompleks II i ubichinol do kompleksu III. Cytochrom c transportuje elektrony z kompleksu III do kompleksu IV. UCP (białko rozprzęgające) pozwala protonom przechodzić przez błonę mitochondrialną bez sprzężenia chemiosmotycznego, tym samym odłączając łańcuch oddechowy od wytwarzania ATP i prowadząc do termogenezy.
1.3. Reaktywne formy tlenu (ROS)
80% tlenu zużywanego przez ssaki jest wykorzystywane w mitochondriach, z czego 1-2% jest przekształcane w reaktywne formy tlenu, tak zwane ROS15. ROS to związki tlenu cząsteczkowego, które są produktem ubocznym metabolizmu komórkowego16. Występują w postaci rodników, jonów lub cząsteczek, które posiadają niesparowany elektron w zewnętrznej powłoce elektronowej17,18. Ze względu na powyższe właściwości, związki te charakteryzują się wysoką reaktywnością i agresywnością chemiczną, a zatem są zdolne do utleniania, a tym samym uszkadzania biopolimerów komórkowych – takich jak DNA, białka i lipidy17-19. Grupa reaktywnych form tlenu obejmuje rodnikowe ROS, takie jak wysoce reaktywny rodnik hydroksylowy (-OH) lub anion ponadtlenkowy (O2•−), który jest uważany za prekursora prawie wszystkich ROS20. Do nierodnikowych ROS zalicza się nadtlenek wodoru (H2O2), wodoronadtlenek (ROOH) lub tlen singletowy (1O2). ROS występują we wszystkich organizmach tlenowych, a ze względu na nieliniową zależność dawka-odpowiedź, przy niskich stężeniach spełniają ważne funkcje fizjologiczne, takie jak transdukcja sygnału w mózgu. W patologicznie wysokich stężeniach ROS przyczyniają się do rozwoju różnych chorób, takich jak stres oksydacyjny16. Mitochondria są uważane za główne źródło tych związków, zwłaszcza mitochondrialnego nadtlenku (mtSO), związku chemicznego zawierającego anion ponadtlenkowy pochodzący z tlenu. Ponieważ ROS i mtSO są niepożądanymi produktami ubocznymi łańcucha oddechowego ( Ryc. 2), powstawanie tych agresywnych związków jest skorelowane z syntezą adenozynotrójfosforanu (ATP) na ostatnim etapie łańcucha oddechowego i zależy od stanu układu oddechowego, tj. gdy potencjał błony mitochondrialnej jest wysoki, a poziom ADP jest niski, produkcja ROS jest wysoka. Gdy potencjał błony jest niski, a poziom ADP wysoki, produkcja ROS jest niska15. Jest to spowodowane ucieczką elektronów między kompleksami I i III18,21,22. Ze względu na stosunkowo wysoką produkcję ROS w mitochondriach oraz fakt, że mtDNA jest bardziej podatne na wpływy zewnętrzne (takie jak ROS) z powodu braku ochronnych białek histonowych, informacja genetyczna tych organeli jest uszkadzana przez oksydację około dziesięć razy częściej niż DNA jądra komórkowego. W tym przypadku wskaźnik mutacji przy braku wpływów zewnętrznych wynosi około jednej wymiany nukleotydów na 1,0 × 109 par zasad w każdej rundzie replikacji15,23. Co więcej, oksydacyjne uszkodzenie mtDNA w łańcuchu oddechowym prowadzi do zwiększenia ucieczki elektronów, a tym samym zwiększenia liczby rodników, które z kolei przyczyniają się do dalszego wzrostu wskaźnika mutacji i uszkodzenia mtDNA24-27. Zgodnie z „mitochondrialną teorią starzenia”, mitochondrialne ROS (mtROS) odgrywają bardzo ważną rolę w procesach starzenia i chorobach związanych z wiekiem wraz z innymi oksydacyjnymi uszkodzeniami mtDNA i ograniczoną zdolnością naprawczą mitochondriów28. Jednak szczegółowe dane, takie jak dokładna ilość mtROS, pozostają kontrowersyjne29,30, chociaż już w latach 80. i 90. wykazano, że ROS poważnie uszkadzają mtDNA31,32.
1.4. Mitochondria w procesie starzenia
Proces starzenia charakteryzuje się wzrostem zaburzeń związanych z wiekiem i poważnymi chorobami. Ze względu na rolę mitochondriów w fosforylacji oksydacyjnej, a tym samym w produkcji ATP, która ma kluczowe znaczenie dla wielu procesów komórkowych, jedną z przyczyn tych zaburzeń i chorób mogą być wadliwe mitochondria30,31. Chociaż starzenie się jest procesem wieloczynnikowym, a do tego znacznie bardziej złożonym niż wcześniej sądzono, w artykule omówione zostaną jedynie mechanizmy mitochondrialne, które odgrywają w nim najważniejszą rolę. W „wolnorodnikowej teorii starzenia” (FRTA), opracowanej przez Harmana w 1956 roku, wolne rodniki zostały zidentyfikowane jako przyczyna procesów starzenia, ponieważ uszkadzają cząsteczki ważne dla wielu ważnych funkcji komórkowych, takie jak DNA, RNA, a także białka i lipidy. Po odkryciu genomu mitochondrialnego Harman rozszerzył swoją teorię do „mitochondrialnej wolnorodnikowej teorii starzenia” (MFRTA), która zajmuje się związaną z wiekiem utratą funkcji mitochondriów z powodu nagromadzenia uszkodzeń oksydacyjnych. Teoria ta znana jest również jako „mitochondrialna teoria starzenia”32,33. Zgodnie z powyższą teorią, nagromadzenie ROS, które uszkadzają mitochondrialne DNA i białka, może prowadzić do dysfunkcji mitochondrialnych w łańcuchu transportu elektronów. Z powodu tych dysfunkcji powstaje więcej ROS, które z kolei uszkadzają mtDNA. Nagromadzenie uszkodzeń mtDNA prowadzi następnie do nowych defektów w łańcuchu transportu elektronów, zwiększonej produkcji ROS i większego stresu oksydacyjnego. Ostatecznie prowadzi to do błędnego koła produkcji ROS24-27. W innych teoriach dotyczących skutków wczesnej dysfunkcji mitochondriów, kluczową rolę odgrywa metabolizm energetyczny. Na przykład „teoria maksymalnego spadku” Prinzingera zajmuje się korelacją między długością życia a produkcją energii w komórkach. Prinzinger twierdzi, że starzenie się jest konsekwencją ograniczonej zdolności mitochondriów do wytwarzania energii34. W innej teorii, modelu wadliwej elektrowni, Krutmann i współpracownicy omawiają rolę mutacji mtDNA i zaburzeń w łańcuchu oddechowym jako czynników dysfunkcji mitochondriów, a tym samym ograniczonej długości życia35. Ostatecznie wszystkie teorie uznają, że proces starzenia jest między innymi konsekwencją dysfunkcji mitochondriów, a organelle te odgrywają bardzo ważną rolę w procesach starzenia i chorobach związanych z wiekiem36-40. Zostało to niedawno potwierdzone w eksperymentach przeprowadzonych z biopsjami ludzkiego naskórka. Po raz pierwszy bezpośrednio w tkance ludzkiej ex vivo zaobserwowano spadek oddychania mitochondrialnego o około 10% na dekadę oraz spadek produkcji ATP wraz ze wzrostem wieku dawcy, co jest zgodne z „mitochondrialną teorią starzenia”41.
2. Dysfunkcje mitochondriów – znaczenie kliniczne
Choroby mitochondrialne, znane również jako mitochondriopatie, to choroby, w których występuje defekt tych organelli. Do tej pory rozpoznano około 50 takich chorób30. Ich przyczyna często znajduje się w jądrze komórkowym i genomie mitochondrialnym. Ze względu na swoją strukturę (brak histonów) i lokalizację w bezpośrednim sąsiedztwie łańcucha oddechowego, mtDNA jest szczególnie podatne na szkodliwe czynniki, takie jak ROS. W rezultacie wskaźnik mutacji w genomie mitochondrialnym jest dziesięciokrotnie wyższy niż w DNA jądra komórkowego. Jednak ze względu na dużą liczbę kopii mtDNA na komórkę, mitochondria – w przeciwieństwie do jądra komórkowego – mają zdolność do eliminowania nawet większych fragmentów wadliwego DNA i zastępowania ich nienaruszonymi kopiami23,42. Jeśli w komórce dojdzie do uszkodzenia około dwóch trzecich wszystkich mitochondriów, prowadzi to do niewydolności mitochondriów i braku odpowiedniego zabezpieczenia komórek w energię. Można się wtedy spodziewać poważnej dysfunkcji mitochondriów oraz związanych z nimi chorób, takich jak cukrzyca, choroby układu krążenia, nowotwory i związane z wiekiem choroby neurodegeneracyjne, takie jak choroba Alzheimera i choroba Parkinsona, które dotyczą głównie komórek o wysokim zapotrzebowaniu na energię, a w konsekwencji większej liczby mitochondriów34,35,42. Początkowe oznaki zmniejszenia liczby nienaruszonych mitochondriów mogą obejmować trudności z pamięcią i koncentracją, spadek sprawności fizycznej oraz pogorszenie wzroku, węchu i słuchu. Tysiące kopii mtDNA w komórce są niemal identyczne w chwili narodzin. Natomiast u pacjentów z dysfunkcją mitochondriów w każdej komórce występuje mieszanka zmutowanego i dzikiego typu mtDNA43,44. Co więcej, zakres mutacji DNA różni się w obrębie narządów i tkanek tego samego osobnika45. Jest to jedno z wyjaśnień dla dużej różnorodności fenotypów u pacjentów z dysfunkcją mitochondriów46. Dotychczas w rozwoju mitochondriopatii skupiano się na dziedziczeniu czynników genetycznych, ale od pewnego czasu uwzględnia się także inne szkodliwe wpływy, takie jak działanie czynników środowiskowych, np. promieniowanie UV28, ale także leki, które znalazły się w centrum uwagi jako czynniki wywołujące dysfunkcje mitochondriów. Zgodnie z teorią endosymbiontów, mitochondria pochodzą od bakterii, które zostały przejęte przez komórki eukariotyczne, więc leki mające na nie szkodliwy wpływ to głównie antybiotyki, ale także chemioterapeutyki i metylofenidat, który między innymi blokuje kompleks I mitochondrialnego łańcucha oddechowego, a tym samym zwiększa produkcję mtSO. W dłuższej perspektywie stan zapalny może również prowadzić do uszkodzenia mitochondriów, ponieważ zawsze towarzyszy mu uwalnianie ROS. Cierpienie psychiczne także prowadzi do powstawania ROS, a tym samym do stresu neurologicznego, który może powodować dysfunkcję mitochondriów47. Obecność wysokiego stężenia ROS lub wzrost stężenia prowadzi do przesunięcia równowagi wewnątrzkomórkowej między utlenianiem a redukcją na korzyść procesów utleniania. Sprzyja to powstawaniu większej ilości ROS i zapobiega skutecznej inaktywacji tych związków przez przeciwutleniacze dostępne dla komórek, co dodatkowo zaburza równowagę19. Zakłócenie wewnątrzkomórkowej równowagi między tworzeniem ROS a ich dezaktywacją nazywane jest stresem oksydacyjnym19,48.
Dla przywrócenia równowagi proponuje się stosowanie substancji mitotropowych, czyli substancji, które pozytywnie wpływają na funkcjonowanie mitochondriów.
Schniertshauer, D.; Wespel, S.; Bergemann, J. Natural Mitochondria Targeting Substances and Their Effect on Cellular Antioxidant System as a Potential Benefit in Mitochondrial Medicine for Prevention and Remediation of Mitochondrial Dysfunctions. Curr. Issues Mol. Biol. 2023, 45, 3911–3932.




Bibliografia
- Klinke, R.; Pape, H.C.; Kurtz, A.; Silbernagl, S. Physiologie, 6th ed.; Georg Thieme Verlag: Stuttgart, Germany, 2009; ISBN 978-3137960065.
- Nordheim, A.; Knippers, R. Molekulare Genetik, 8th ed.; Georg Thieme Verlag: Stuttgart, Germany, 2018; ISBN 978-3132426375.
- Schatz, G. The Magic Garden. Annu. Rev. Biochem. 2007, 76, 673–678.
- Zorov, D.B.; Krasnikov, B.F.; Kuzminova, A.E.; Vysokikh, M.Y.; Zorova, L.D. Mitochondria Revisited. Alternative Functions of Mitochondria. Biosci. Rep. 1997, 17, 507–520.
- Alberts, B. Lehrbuch der Molekularen Zellbiologie, 5th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2015; ISBN 978-3-527-32384-5.
- Madigan, M.T.; Martinko, J.M.; Stahl, D.A.; Clark, D.P. Brock Mikrobiologie, 13th ed.; Pearson: München, Germany, 2013; ISBN 978-3868941449.
- Adrain, C.; Martin, S.J. The mitochondrial apoptosome: A killer unleashed by the cytochrome seas. Trends Biochem. Sci. 2001, 26, 390–397.
- Gvozdjáková, A. Mitochondrial Medicine: Mitochondrial Metabolism, Diseases, Diagnosis and Therapy, 2008th ed.; Springer: Berlin, Germany, 2008; ISBN 978-1402067136.
- Lill, R.; Diekert, K.; Kaut, A.; Lange, H.; Pelzer, W.; Prohl, C.; Kispal, G. The Essential Role of Mitochondria in the Biogenesis of Cellular Iron-Sulfur Proteins. Biol. Chem. 1999, 380, 1157–1166.
- DiMauro, S.; Schon, E.A. Mitochondrial DNA mutations in human disease. Am. J. Med. Genet. 2001, 106, 18–26.
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630.
- Meinecke, M.; Wagner, R.; Kovermann, P.; Guiard, B.; Mick, D.U.; Hutu, D.P.; Voos, W.; Truscott, K.N.; Chacinska, A.; Pfanner, N.; et al. Tim50 Maintains the Permeability Barrier of the Mitochondrial Inner Membrane. Science 2006, 312, 1523–1526.
- Nelson, D.; Cox, M. Lehninger Biochemie, 2011st ed.; Springer: Berlin, Germany, 2011; ISBN 978-3540686378.
- Nedergaard, J.; Ricquier, D.; Kozak, L.P. Uncoupling proteins: Current status and therapeutic prospects. EMBO Rep. 2005, 6, 917–921.
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199.
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12, Erratum in Mol. Cell. Biochem. 2020, 467, 13.
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583.
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496.
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allerg. Organ. J. 2012, 5, 9–19.
- Lambert, A.J.; Brand, M.D. Reactive Oxygen Species Production by Mitochondria. Methods Mol. Biol. 2009, 554, 165–181.
- Bottje, W. Oxidative metabolism and efficiency: The delicate balancing act of mitochondria. Poult. Sci. 2019, 98, 4223–4230.
- Hunte, C.; Zickermann, V.; Brandt, U. Functional Modules and Structural Basis of Conformational Coupling in Mitochondrial Complex I. Science 2010, 329, 448–451.
- Wirth, C.J.; Zichner, L. Orthopädie und Orthopädische Chirurgie: Stoffwechel-und Systemerkrankungen, 1st ed.; Georg Thieme Verlag: Stuttgart, Germany, 2003; ISBN 9783131269317.
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692.
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.-S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free. Radic. Res. 2012, 46, 147–153.
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187.
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagen- esis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104.
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Einfluss von Solarer Strahlung auf die Mitochondrien. OM & Ernährung. 2018, SH08, pp. 50–57. Available online: https://www.omundernaehrung.com/einfluss-von-solarer-strahlung-auf-die-mitochondrien. html (accessed on 19 October 2022).
- Lagouge, M.; Larsson, N. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543.
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial production of reactive oxygen species. Biochemistry 2013, 78, 1490–1511.
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147.
- Prinzinger, R. Programmed ageing: The theory of maximal metabolic scope. How does the biological clock tick? EMBO Rep. 2005, 6 (Suppl. 1), S14–S19.
- Krutmann, J.; Schroeder, P. Role of Mitochondria in Photoaging of Human Skin: The Defective Powerhouse Model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49.
- Damian, M.S.; Ellenberg, D.; Gildemeister, R.; Lauermann, J.; Simonis, G.; Sauter, W.; Georgi, C. Coenzyme Q10 Combined With Mild Hypothermia After Cardiac Arrest: A preliminary study. Circulation 2004, 110, 3011–3016.
- Hazane, F.; Sauvaigo, S.; Douki, T.; Favier, A.; Beani, J.-C. Age-dependent DNA repair and cell cycle distribution of human skin fibroblasts in response to UVA irradiation. J. Photochem. Photobiol. B 2006, 82, 214–223.
- Prahl, S.; Kueper, T.; Biernoth, T.; Wöhrmann, Y.; Münster, A.; Fürstenau, M.; Schmidt, M.; Schulze, C.; Wittern, K.-P.; Wenck, H.; et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. Biofactors 2008, 32, 245–255.
- Sauvaigo, S.; Bonnet-Duquennoy, M.; Odin, F.; Hazane-Puch, F.; Lachmann, N.; Bonté, F.; Kurfürst, R.; Favier, A. DNA repair capacities of cutaneous fibroblasts: Effect of sun exposure, age and smoking on response to an acute oxidative stress. Br. J. Dermatol. 2007, 157, 26–32.
- Wei, Y.-H.; Wu, S.-B.; Ma, Y.-S.; Lee, H.-C. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang Gung Med. J. 2009, 32, 113–132.
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Age-Dependent Loss of Mitochondrial Function in Epithelial Tissue Can Be Reversed by Coenzyme Q10. J. Aging Res. 2018, 2018, 6354680.
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79.
- Holt, J.I.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719.
- Holt, I.J.; Harding, E.A.; Petty, R.K.; Morgan-Hughes, A.J. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433.
- Macmillan, C.; Lach, B.; Shoubridge, E.A. Variable distribution of mutant mitochondria1 DNAs (tRNALeu[3243]) in tissues of symptomatic relatives with MELAS: The role of mitotic segregation. Neurology 1993, 43, 1586.
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141.
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.